

Da wir vor einer Weile das Thema „Arzneientwicklung“ mal im Chat angesprochen hatten, dachte ich mir ich schreibe dazu mal etwas ausführlicher, wie das ganze genau abläuft.
Es gibt grundsätzlich einmal ein recht einheitliches internationales Schema, welchem die meisten Länder (vor allem Japan, die EU und Nordamerika) für die Entwicklung und Vermarktung folgen und so auch von den verantwortlichen Behörden (Etwa die FDA oder EMA) gehandhabt wird.

Dazu hat jedes Land meist noch so ein bisschen seine eignen regulatorischen Feinheiten, aber der Einfachheit halber bleibe ich mal beim Schema wie oben abgebildet.
Wie man am Bild schon ablesen kann, ist es keine sehr günstige Sache ein Medikament auf den Markt zu bringen und dauern tut es ebenfalls. Das mag auch zwischen den unterschiedlichen Kategorien variieren, ein Hustensäftle ist da etwas leichter zu handhaben als etwa eine neue Krebstherapie oder Impfung. Je heikler und schwerwiegender, desto höher an sich auch die Kosten und benötigte Zeit, da mehr Testhürden und Auflagen für Sicherheit von Patienten zu erfüllen sind (als Faustregel).
Mal abgesehen von der üblichen Konzernprofitgier, die auf den Preis schlägt, ist bei der Branche auch die hohe Ausfallrate an potenziellen Substanzen das Problem. Wir ihr auch oben im Bild sehen könnt, kommt aus der Forschungsphase eine ganze Bandbreite an Kandidaten, die dann über die weiteren Testphasen immer weniger werden.
Das Ausscheiden kann unterschiedliche Gründe haben, von Herstellungsschwierigkeiten über Giftigkeit bis hin zur schnelleren Konkurrenz oder dem eignen Bankrott kann alles zur Terminierung eines Kandidaten führen. Das derweil allerdings mehrere Millionen bis Milliarden in einen gescheiterten Versuch rein fließen können, muss am Ende der eine, der es schafft die Verluste durch die anderen 9999 ausgleichen und nebenbei auch noch etwas Gewinn abwerfen. Durchschnitt sind aber wie im Bildchen oben meist 10-12 Jahre und etwa knapp ne Milliarde Euro an Gesamtkosten
Forschungsphase
Gut, genug von Kosten und Zeitfressern, wie läuft das ganze nun eigentlich ab? An sich wird recht selten ins blaue Hinein geforscht, viel eher wird davor schon einmal ein Ziel festgesteckt. Etwa, man möchte ein neues Kopfschmerzmittel, am besten als Tröpfen verabreichbar. Man steckt also ab soweit man kann, welches Ziel das neue Produkt haben soll, wie es am besten angewandt wird und dergleichen. Das kann am Anfang noch etwas nebulös sein, wird aber über die Dauer des Projekts und konkreteren Testresultaten mit der Zeit immer mehr fixiert. Welche Dosis, welche Form, welcher Name, welche Zielgruppe, etc.
Gut, hat man nun ein Ziel, so wird in der Forschungsabteilung erst einmal kräftig gewerkelt, da vergehen meist einmal 2-3 Jahre oder auch ein bisschen mehr, am Ende steht man dann meist mit ein paar tausend Kandidaten da, die in etwa das tun, was man von ihnen erwartet. In unserem Beispiel etwa Kopfschmerzen eindämmen.
Vorklinische Phase
Jetzt ist die Frage, welche davon auch wirklich in Menschen funktionieren, wie viel man davon sicher anwenden kann und wie es um die Herstellung und Formulierung bestellt ist. Diese Fragen versucht man nun in der vorklinischen Phase auszubügeln, in der Zeit gehen auch die meisten Kandidaten flöten (hier ist auch der bisherige Kostenaufwand noch nicht so hoch). In der Phase wird die Sicherheit und ungefähre sichere Dosierungsspannweite erkundet, dabei wird sich mit den verbleibenden Kandidaten über eine immer komplexer werdende Kaskade von Versuchen an ein akzeptables Ergebnis herangetastet.
In der Praxis sieht das so aus, dass oft erst einmal in einfachen Zellkulturen angefangen wird (da scheidet der Großteil aus) und von dort dann über komplexere Zellkulturen und/oder „niedere“ Tiere (etwa Zebrafisch) immer näher an das menschliche Model herangetastet wird. Je ähnlicher desto besser, da es letztendlich logischerweise auch mehr Schlussfolgerungen zulässt, wenn der getestete Organismus dem Menschen ähnlicher ist. Gleichzeitig sind die ähnlicheren Organismen aber meist auch schwieriger zu testen und zu halten und bedürfen meist auch schwieriger behördlicher und ethischer Auflagen, weshalb erst mit einfacheren Tieren angefangen wird, bevor es kniffliger wird.
Als Beispiel: Zellkultur > Zebrafisch > Maus > Hund > Affe
Klinische Phasen
Haben es die wenigen verbleibenden Kandidaten über die vorklinische Hürde geschafft, kommt der kritische Teil, nämlich die klinischen Studien. Dabei gibt es unterschiedliche Phasen, hier mal in dem Bild schön zusammengefasst
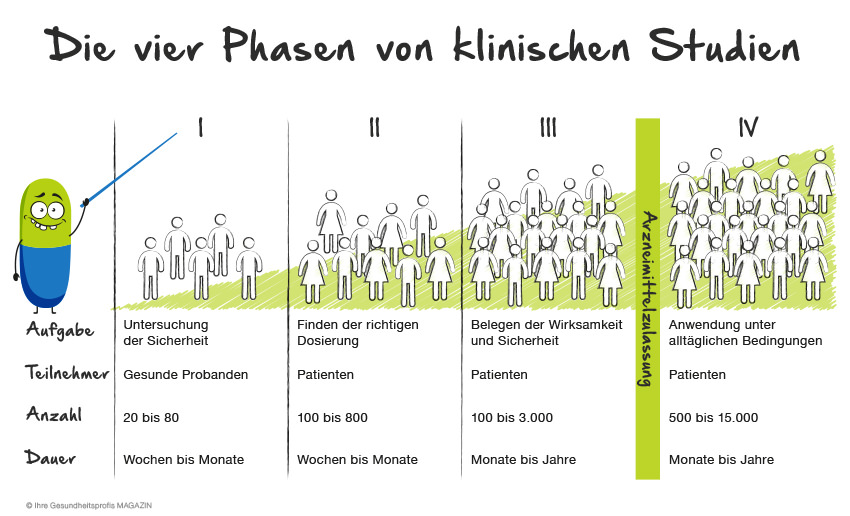
Für die Zulassung eines Stoffes bedarf es wiederum der Zustimmung einer Ethikkomission. Die wiederum kriegt man nicht ohne das man minimale Risikofaktoren und wirklichen Bedarf für die Substanz begründen kann, deshalb auch die exzessiven vorklinischen Tests als Rechtfertigungsgrundlage.
Phase I
Hat man die Erlaubnis nun bekommen, geht es ans Phase I
In den vorklinischen tests wurde die ungefähre Dosierungsspanne untersucht und die Giftigkeit zu ergründen, da sonst das Risiko des Abnippels für die Probanden zu groß wäre. Darum auch die Tests an möglichst menschenähnlichen Tieren, um das Risiko so gering wie möglich zu halten. In Phase 1 wird generell die Sicherheit für Menschen erkundet, dazu werden hierfür auch nur gesunde Freiwillige zugelassen und die Anzahl der Probanden ist auch sehr klein. Mehr als ein paar dutzend sind das meist nicht.
Ausnahme hierfür sind schwere Geschütze wie Arznei für Chemotherapie. Da man grundsätzlich schon einmal davon ausgehen kann, dass das für den menschlichen Körper kein leicht zu handelndes Erlebnis ist, wird hierfür meist gleich mit Patienten begonnen, die etwa schon ein weit fortgeschrittenes Krankheitsstadium haben. Auch hier ist die Anzahl der Personen allerdings meist sehr überschaubar.
In dieser ersten Phase wird meist dann nochmal der Großteil der Kandidaten eliminiert, wenn sie als zu unsicher eingestuft werden, am Ende hat man dann oft nicht einmal mehr ein halbes Dutzend.
Phase II & Phase III
In Phase II und III wird dann mit größeren Probandenzahlen gearbeitet, wobei es hier nun ausschließlich um Patienten handelt. In diesen beiden Phasen wird versucht die optimale Dosis zu ermitteln, also die Balance zwischen besten Wirkungsgrad und möglichst niedrigen Nebenwirkungen zu ermitteln sowie dies möglich an einer großen Bandbreite verschiedener Patiententypen abzuklären. Geschlecht, Alter, Lebensstil, Ethnie (=genetischer Hintergrund), Statur, all das sind Faktoren die einen Einfluss haben können. Deshalb wird auch versucht möglichst viele Patienten innerhalb dieser Phasen abzudecken, um entsprechende Daten zu sammeln.
Authorisierung
Nach Phase III ist dann von der verbliebenen Handvoll meist nur noch 1-2 übrig, die erfolgreich eine Zulassung erhalten. Danach ist der Spaß aber noch nicht vorbei, es kommt vor, dass eine Phase IV noch verlangt wird, wobei etwa mögliche Nebenwirkungen die im Zusammenhang stehen könnten weiterhin dokumentiert und in die Datensammlung miteinbezogen werden. Sprich, wenn unser Kopfwehbeispiel verschrieben wird und der Patient 2 Tage später erneut beim Doktor landet, weil er einen Ausschlag hat, dann wird dies (sofern noch nicht bekannt), dokumentiert und gegebenenfalls untersucht. Dabei kann es auch schon einmal vorkommen, dass wegen eines in den Phase II und III nicht ermittelten Nebeneffekts ein Medikament für eine bestimmte Patientengruppe oder gar komplett seine Autorisierung verliert, wie das damals bei Kontagan der Fall war.
Am Ende noch kurz was zu ein paar wichtigen Punkten. Sicherheit in sich ist für ein Produkt, was Leben retten oder zumindest erleichtern soll, oberstes Gebot. Dabei gibt es allerdings eine Faustregel, nämlich, dass es kein Medikament ohne (potenzielle) Nebenwirkungen gibt (von Lachen jetzt vielleicht mal abgesehen).
Sprich man muss versuchen möglichst eine Dosierung mit maximaler Effizienz und minimalen vertretbaren Nebenwirkungen zu erreichen. Eine Arznei sollte auch logischerweise keine häufigen schwerwiegenderen Nebenwirkungen haben als die Krankheit, gegen die sie helfen soll.
Ein Kopfschmerzmittel, von dem einem der Arm abfault wäre eher nicht wirklich brauchbar.
Ein anderes Problem sind sehr seltene Krankheiten, für die sich nur sehr schwer Patienten finden lassen. Das macht es zum einen schwierig überhaupt wen zu finden, der etwas dafür entwickelt, weil klarerweise die Anwendermenge zu gering ist um sich zu lohnen. Und zum anderen auch, da es zu wenig Personen für die klinischen Studien gibt. Das ist etwa aktuell bei einer Behandlung gegen Ebola ein Problem. Da sich die Epidemie mittlerweile ausgebrannt hat, gibt es keine Testpersonen mehr für die klinischen Testphasen, weshalb das nun alles im Sand verläuft…
Hmm, soweit ein kleiner Einblick in die Branche meinerseits, viel Spaß den Interessierten beim Lesen.
Ist natürlich nur sehr oberflächlich gehalten und in die einzelnen Punkte wie Studiendesign und Ethikkomissionen ließe sich noch um einiges tiefer eintauchen. Dazu schreibe ich zu einem späteren zeitpunkt vielleicht noch was
Khezef


